KIK-AS. Phase 1/2 Clinical Trial
Note from the editor: This article represents the presentation during the webinar. YouTube recording may differ.

Disclaimer
We’re talking about a trial, and I know I repeat myself, but it’s so important. A trial is a trial, and the aim of this trial is to answer a question, it’s not to deliver a treatment or a cure. When we come into a trial as an investigator, we don’t come to treat your child, it’s because we want to answer a very specific question: Is it safe and does it work? And today we don’t have the answer to this question. I want to make it very clear because if someone tells you one day, “Come include your child in this trial, this is important, and this will help and this is safe and this is efficient”, then this person is lying to you. An honest investigator will start by explaining to you that if we conduct the trial, it’s to assess the safety and efficacy and that we don’t have the answer before running the trial.
So I’m going to show you in this presentation some slides that have been provided to me by the sponsor. You will hear about very early findings from the initial trial. This is very early, there was a very limited number of patients. So we must keep our feet on the ground to appreciate this data.
The molecule
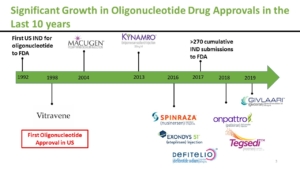 The molecule we’re going to speak about is an oligonucleotide and it’s not a brand-new type of medication. There are already several drugs that have been approved in this family of drugs. The first one was approved, believe it or not, 23 years ago. And now we have quite an important experience with another intrathecal antisense, which is Spinraza in Spinal Muscular Atrophy, where more than 10,000 children and adults have been injected so far.
The molecule we’re going to speak about is an oligonucleotide and it’s not a brand-new type of medication. There are already several drugs that have been approved in this family of drugs. The first one was approved, believe it or not, 23 years ago. And now we have quite an important experience with another intrathecal antisense, which is Spinraza in Spinal Muscular Atrophy, where more than 10,000 children and adults have been injected so far.
 This molecule that we’re using is something that looks like an RNA, but it is not exactly an RNA. It’s more robust, and it helps to alter the expression of the RNA. I won’t go again into the mechanism of action because Dora explained it probably better than I’m able to do.
This molecule that we’re using is something that looks like an RNA, but it is not exactly an RNA. It’s more robust, and it helps to alter the expression of the RNA. I won’t go again into the mechanism of action because Dora explained it probably better than I’m able to do.
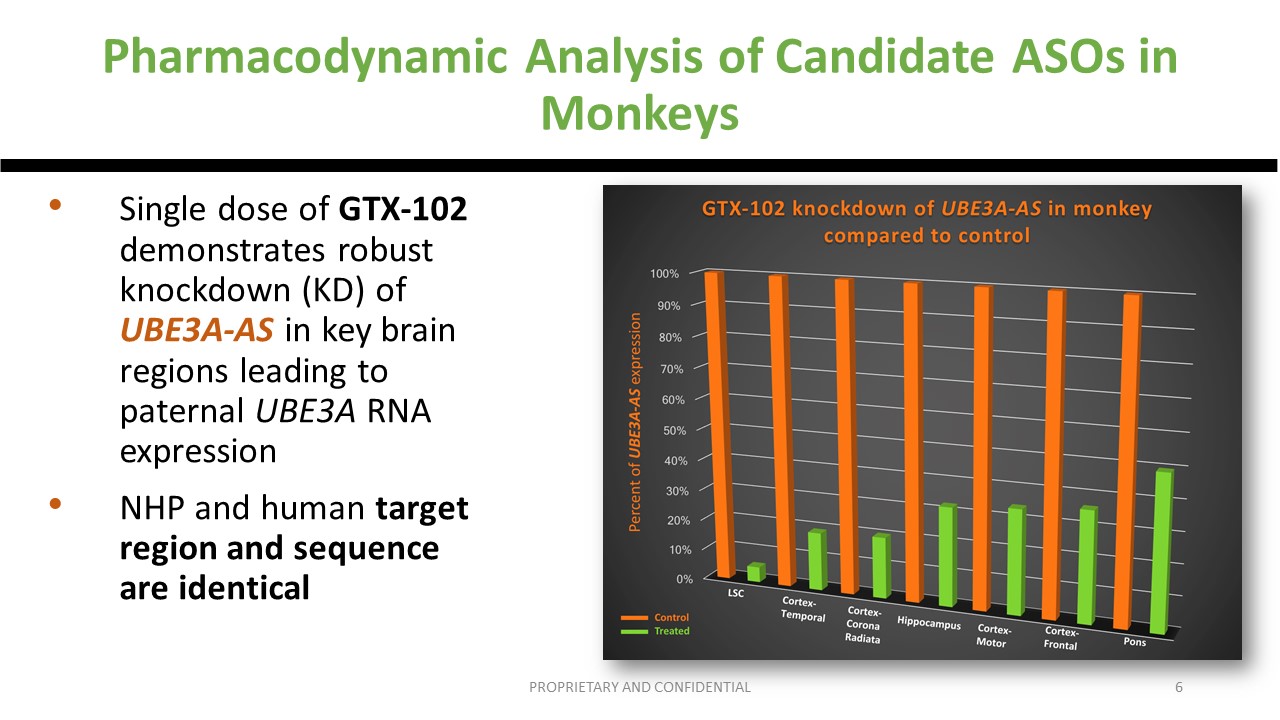
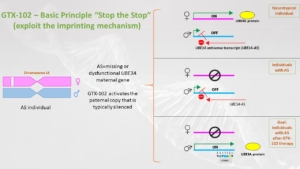 Just to remind, before going to humans, whether we like it or not, there are animal studies, the non-human primate studies, in which we could find that the expression, the pharmacokinetics of the drug, is acceptable and that the regions of the brain that need to be targeted are actually targeted with a knockdown of the UBE3A-AS. And it led to the first Phase 1/2 study that happened in the US.
Just to remind, before going to humans, whether we like it or not, there are animal studies, the non-human primate studies, in which we could find that the expression, the pharmacokinetics of the drug, is acceptable and that the regions of the brain that need to be targeted are actually targeted with a knockdown of the UBE3A-AS. And it led to the first Phase 1/2 study that happened in the US.

GTX-102 Phase 1/2 Study in the US
Study Design
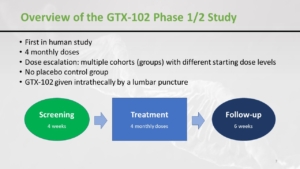 There was no placebo used in the US study. And I just also wanted to make things clear. We’re not objective, I’m not objective. And the reason why is because I like my patients, so I want them to be better. That’s why in studies at some point we need a placebo. At the end of the day, we conduct a trial because we want to have answers and to have answers that we trust. And to trust them at one point we need to be blinded: the parents and the doctors. Sometimes, in the very early phase, and this is the case in this study, there is a placebo. But then it means that we must be extremely cautious with the data in terms of efficacy. And maybe one day with some of you we will discuss a placebo-controlled study. This is important, the aim of all of this is to bring a safe and efficient treatment onto the market, not an inefficient treatment. We don’t care about inefficient treatments, and for that, at some point, we need confirmatory trials with placebo. But this very trial has no placebo, which means that we must be very cautious when we speak about the efficacy.
There was no placebo used in the US study. And I just also wanted to make things clear. We’re not objective, I’m not objective. And the reason why is because I like my patients, so I want them to be better. That’s why in studies at some point we need a placebo. At the end of the day, we conduct a trial because we want to have answers and to have answers that we trust. And to trust them at one point we need to be blinded: the parents and the doctors. Sometimes, in the very early phase, and this is the case in this study, there is a placebo. But then it means that we must be extremely cautious with the data in terms of efficacy. And maybe one day with some of you we will discuss a placebo-controlled study. This is important, the aim of all of this is to bring a safe and efficient treatment onto the market, not an inefficient treatment. We don’t care about inefficient treatments, and for that, at some point, we need confirmatory trials with placebo. But this very trial has no placebo, which means that we must be very cautious when we speak about the efficacy.
 The primary objective, the reason why we conduct the trial, in Phase 1 is safety. And then pharmacokinetics while the efficacy is exploratory. And that’s exactly the hierarchy of the objectives of a study.
The primary objective, the reason why we conduct the trial, in Phase 1 is safety. And then pharmacokinetics while the efficacy is exploratory. And that’s exactly the hierarchy of the objectives of a study.
 The key inclusion criteria of the trial, which are exactly the same as in the US, are as follows. Firstly, it’s the deletion, not a point mutation or UPD. And it’s patients between 4 and 17 years old. Stable seizures control, normal kidney and liver tests and an ability to tolerate anaesthesia. As you can imagine, an intrathecal injection in an Angelman Syndrome patient is not possible without sedation or anaesthesia.
The key inclusion criteria of the trial, which are exactly the same as in the US, are as follows. Firstly, it’s the deletion, not a point mutation or UPD. And it’s patients between 4 and 17 years old. Stable seizures control, normal kidney and liver tests and an ability to tolerate anaesthesia. As you can imagine, an intrathecal injection in an Angelman Syndrome patient is not possible without sedation or anaesthesia.
Does it mean that the drug cannot work in patients with a mutation? Of course not. Does it mean that the drug would not work in an 18 years old? Of course not. But when you start development, you need to be restrictive and then you expand.
 So there are inclusion criteria and exclusion criteria. Patients who would require intubation for injection are not included. Patients with bleeding or a platelet disorder – because of the intrathecal injection. And it has to be an ambulant patient. Of course, it doesn’t mean that the drug won’t work in non-ambulant patients, but at one point you need to have a clear view and a clear outcome.
So there are inclusion criteria and exclusion criteria. Patients who would require intubation for injection are not included. Patients with bleeding or a platelet disorder – because of the intrathecal injection. And it has to be an ambulant patient. Of course, it doesn’t mean that the drug won’t work in non-ambulant patients, but at one point you need to have a clear view and a clear outcome.
 There are a lot of assessments involved and nobody can say that this is an easy trial when you just receive the drug and then you are just doing better. No, there are plenty of assessments. And the reason why, and you know it better than me, is that in Angelman Syndrome there is a lot of aspects about speech, seizures, sleep, behaviour, ambulation, etc. And there is not a single outcome that can encompass all these aspects. And this can be burdensome, but we want to know what happened to the children. And we want to know if the drug has some effect on the different components of the syndrome.
There are a lot of assessments involved and nobody can say that this is an easy trial when you just receive the drug and then you are just doing better. No, there are plenty of assessments. And the reason why, and you know it better than me, is that in Angelman Syndrome there is a lot of aspects about speech, seizures, sleep, behaviour, ambulation, etc. And there is not a single outcome that can encompass all these aspects. And this can be burdensome, but we want to know what happened to the children. And we want to know if the drug has some effect on the different components of the syndrome.
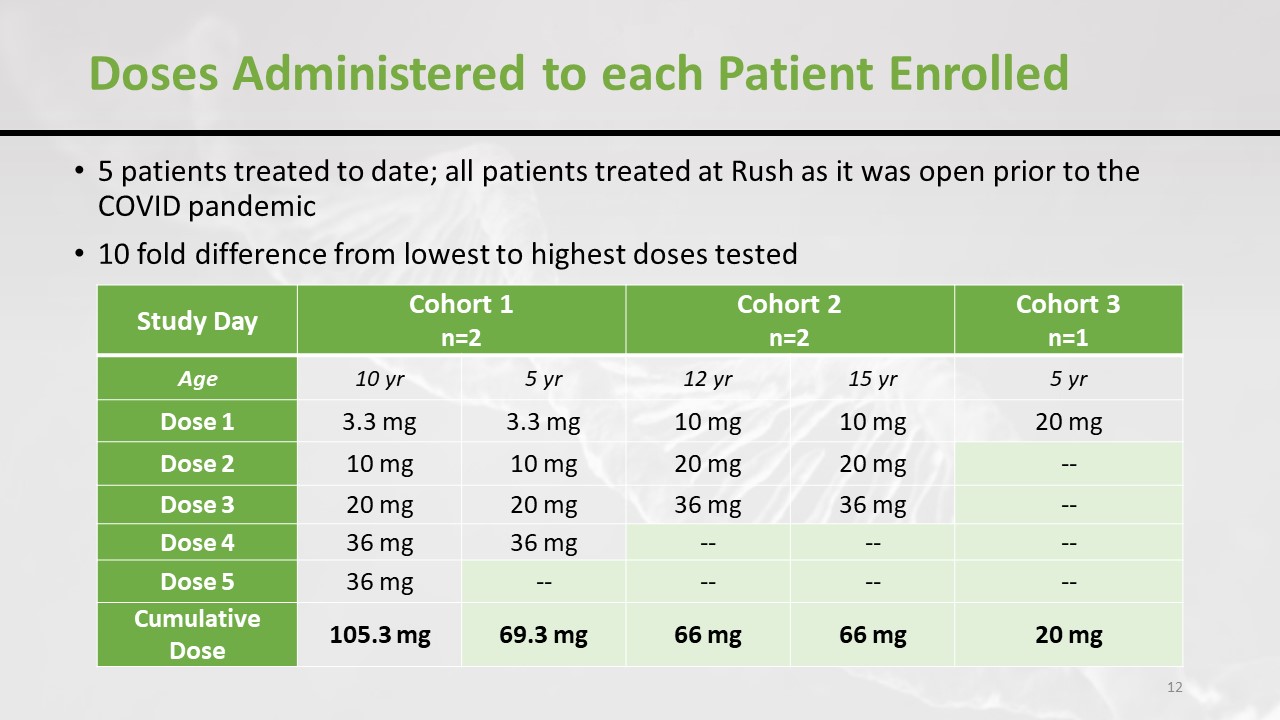
 What happened in the US trial is that there were five patients included. And, as you might imagine, it was a little bit disrupted by the pandemic, and there were 3 cohorts. And as you can see, the starting dose was higher in the last cohort. So they increased the dose progressively and here’s the total accumulated dose that the children have received.
What happened in the US trial is that there were five patients included. And, as you might imagine, it was a little bit disrupted by the pandemic, and there were 3 cohorts. And as you can see, the starting dose was higher in the last cohort. So they increased the dose progressively and here’s the total accumulated dose that the children have received.
Safety
 What happened in terms of safety, and one more time – safety was the primary objective, is that there was an increase of ataxia and fatigue after the dose. It lasted 1 to 3 days and started 2 to 6 hours after the injection. Just really related to the injection. And the severity was very different from one patient to another. And then, of course, there were symptoms associated with a lumbar puncture, like a headache. But, overall, the anaesthesia and the lumbar puncture were quite well tolerated.
What happened in terms of safety, and one more time – safety was the primary objective, is that there was an increase of ataxia and fatigue after the dose. It lasted 1 to 3 days and started 2 to 6 hours after the injection. Just really related to the injection. And the severity was very different from one patient to another. And then, of course, there were symptoms associated with a lumbar puncture, like a headache. But, overall, the anaesthesia and the lumbar puncture were quite well tolerated.
 What happened is that at the highest dose cohort the patients presented a lower extremity weakness. And in two patients it was quite severe. The good news is that it recovered after the stop of treatment, but as you can imagine if this kind of effect happens with a high dose you cannot continue with a high dose. You have to downgrade, stop the study to make an amendment and go to a lower dose that doesn’t cause this lower extremity weakness in the patients. The good news is that it fully recovered, and that’s also why we conduct trials. It’s because we know that the follow up in the trial is much different than what happened in the real life that we need to interrupt things when they get out of hand.
What happened is that at the highest dose cohort the patients presented a lower extremity weakness. And in two patients it was quite severe. The good news is that it recovered after the stop of treatment, but as you can imagine if this kind of effect happens with a high dose you cannot continue with a high dose. You have to downgrade, stop the study to make an amendment and go to a lower dose that doesn’t cause this lower extremity weakness in the patients. The good news is that it fully recovered, and that’s also why we conduct trials. It’s because we know that the follow up in the trial is much different than what happened in the real life that we need to interrupt things when they get out of hand.
 We could understand that this weakness was related to an inflammation of the nerves that responsible for the movement of the legs. That was a real safety issue and that’s why the trial was put on hold. Because we needed an amendment to restart with a lower dose.
We could understand that this weakness was related to an inflammation of the nerves that responsible for the movement of the legs. That was a real safety issue and that’s why the trial was put on hold. Because we needed an amendment to restart with a lower dose.
Efficacy
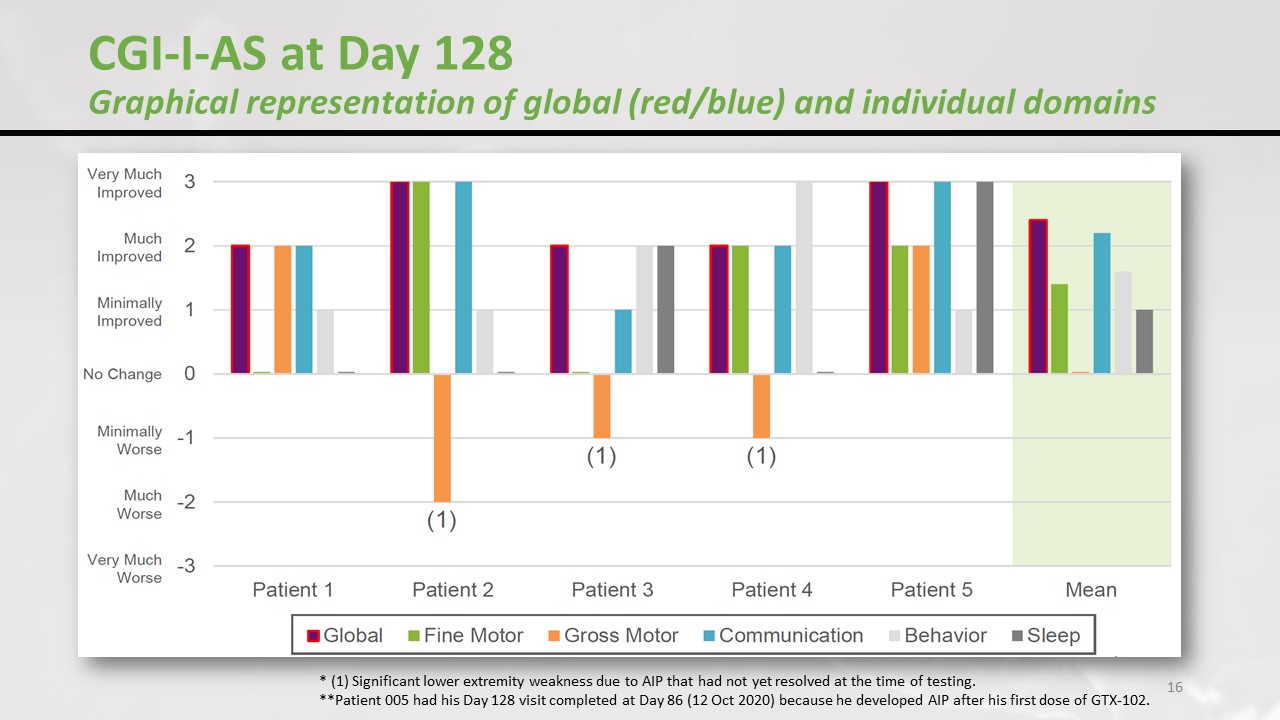
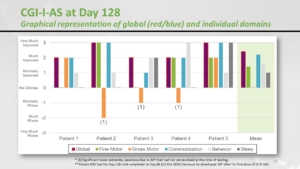 When we ask the parents, in terms of speech, fine motor skills, communication, behaviour, sleep, how would you say your child is doing? No change / minimally improved / much improved / very much. That’s how the parents ranged the children. The motor perspective in 3 patients went down, those were the patients who developed a serious adverse reaction – the weakness in the legs. But if we consider the other aspects then it was quite efficient. But one more time, let’s remind you this is not a double-blind randomised placebo-controlled study. The parents know that their child is on medication and they are probably biassed. This was a nice signal of efficacy but we must keep our feet on the ground.
When we ask the parents, in terms of speech, fine motor skills, communication, behaviour, sleep, how would you say your child is doing? No change / minimally improved / much improved / very much. That’s how the parents ranged the children. The motor perspective in 3 patients went down, those were the patients who developed a serious adverse reaction – the weakness in the legs. But if we consider the other aspects then it was quite efficient. But one more time, let’s remind you this is not a double-blind randomised placebo-controlled study. The parents know that their child is on medication and they are probably biassed. This was a nice signal of efficacy but we must keep our feet on the ground.
Comment from Scott Stromatt, chief medical officer for GeneTX: “I just want to clarify for the group a minor point that the CGI was actually completed by the physician investigator. So the investigator examined the patient (physical and neurologic exam), took the information, all the clinical information interviewing the parents. And it was a gestalt rating, their rating of how the child was doing.”
UK Trial
 So, as you might imagine when a safety concern arises we don’t want to continue with a very high dose. We have to restart with a lower dose. And so now in the amendment patients from 4 to 8 will start from 3.3mg and then increase progressively to a maximum of 14mg, unlike the previous study where it was 36mg.
So, as you might imagine when a safety concern arises we don’t want to continue with a very high dose. We have to restart with a lower dose. And so now in the amendment patients from 4 to 8 will start from 3.3mg and then increase progressively to a maximum of 14mg, unlike the previous study where it was 36mg.

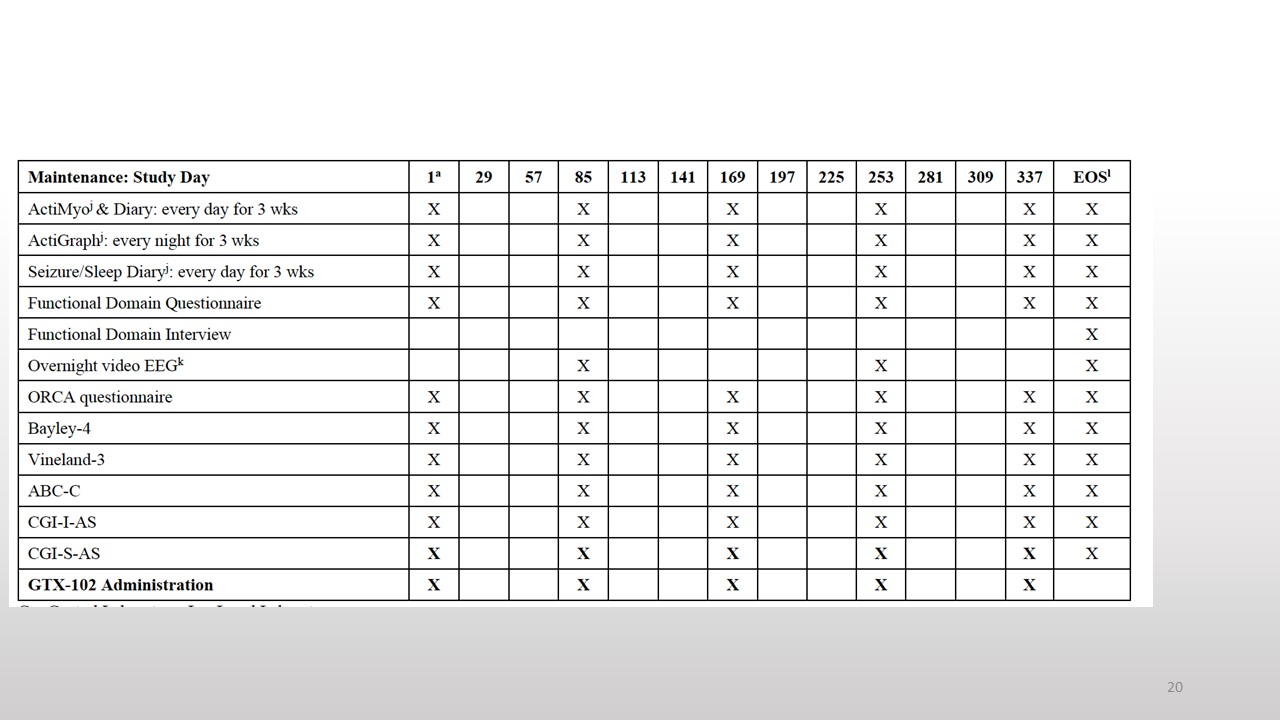
 Nobody can say that there is nothing to do in this trial. You can see there is a screening period, then there is a baseline. Later patients come for an injection to stay in the hospital for 24 hours. This is followed by the dose-escalation phase. And a follow-up and if there is some benefit the program continues and the maintenance period is every 84 days or 12 weeks.
Nobody can say that there is nothing to do in this trial. You can see there is a screening period, then there is a baseline. Later patients come for an injection to stay in the hospital for 24 hours. This is followed by the dose-escalation phase. And a follow-up and if there is some benefit the program continues and the maintenance period is every 84 days or 12 weeks.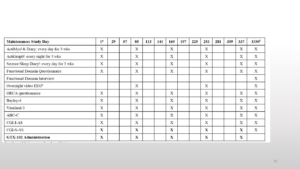
About anaesthesia. We cannot compare with Spinal Muscular Atrophy because there is no cognitive impairment in Spinal Muscular Atrophy. There is a paper recently published that explains how they managed the anaesthesia. They used a simple inhalation induction and mask maintenance, and it was successful with 1.3% major adverse events and 4.8% minor adverse events as in any anaesthesia.
 So there is information posted on a regular basis on ClinicalTrials.gov. The name of the trial, believe it or not, is “KIK-AS”, and there’s a website KIK-AS.com. Do not hesitate to discuss with your GP and also with the patients’ advocacy groups FAST UK and AngelmanUK.
So there is information posted on a regular basis on ClinicalTrials.gov. The name of the trial, believe it or not, is “KIK-AS”, and there’s a website KIK-AS.com. Do not hesitate to discuss with your GP and also with the patients’ advocacy groups FAST UK and AngelmanUK.
 I’ve put up my address, and if you want to discuss this trial, and if you want your child to be considered, feel free to drop me an email. I cannot contact you, but you can contact me. By the way, the same applies to the Natural History Study. If you’re convinced as we are that this is the way to move just contact us.
I’ve put up my address, and if you want to discuss this trial, and if you want your child to be considered, feel free to drop me an email. I cannot contact you, but you can contact me. By the way, the same applies to the Natural History Study. If you’re convinced as we are that this is the way to move just contact us.
Spinal Muscular Atrophy Pledge
 As you are all here, I’m going just to advocate for another disease – SMA, which is Spinal Muscular Atrophy. We have drugs that work, and I truly hope that one day we will have one or more drugs for Angelman Syndrome as we do for SMA. And that day we will need the newborn screening. Today it’s too early for Angelman Syndrome, but for SMA we need the newborn screening now. This is crucially important for patients to come and patients who are already born.
As you are all here, I’m going just to advocate for another disease – SMA, which is Spinal Muscular Atrophy. We have drugs that work, and I truly hope that one day we will have one or more drugs for Angelman Syndrome as we do for SMA. And that day we will need the newborn screening. Today it’s too early for Angelman Syndrome, but for SMA we need the newborn screening now. This is crucially important for patients to come and patients who are already born.
So please, if you want to help the rare disease community and to show the government that we collectively care about rare diseases, please take the time to look at the parliament petition in please sign it. Today we have more than 7000 signatures after a couple of days. If you believe that it’s the right thing to do, not only sign it, also share it on Facebook with your network. Let’s show the government that we care about rare diseases and about the patients in this country.
My biggest hope is that in 5 or 10 years, I’ll say please sign the petition for the newborn screening for Angelman Syndrome. And this will happen if we are clever enough to tick the boxes to bring safe and efficient drugs on the market. Thank you for your attention.